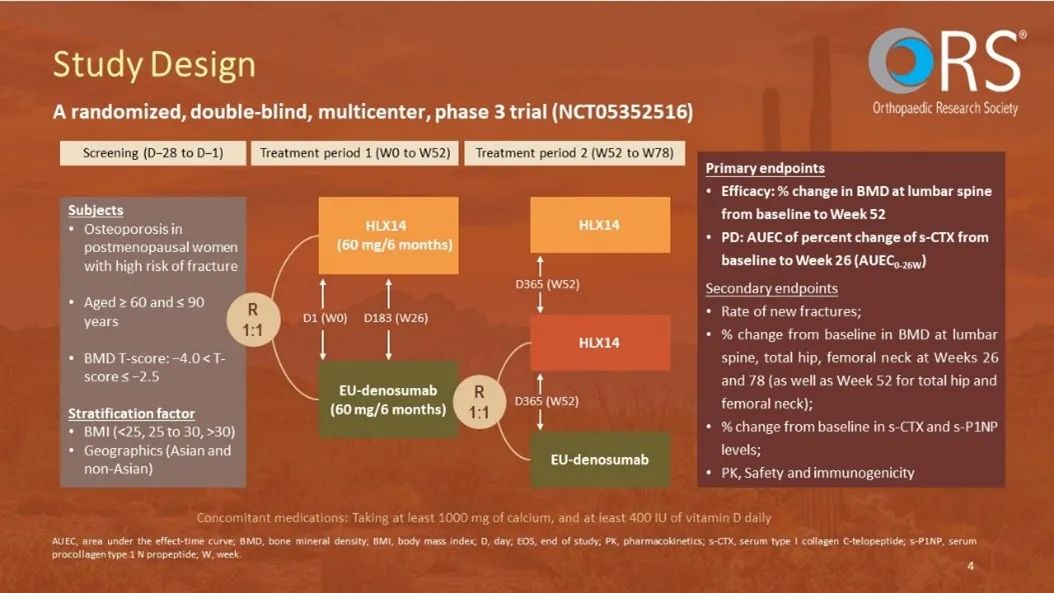
试验设计: 本研究为一项随机、双盲、国际、平行对照的III期研究。受试者为年龄在60至90岁之间、确认已绝经且腰椎或全髋骨矿物质密度(BMD)T评分通过中心双能X线吸收法扫描为−4.0 < T评分 ≤ −2.5的女性。合格的受试者按1:1分配分别接受两剂HLX14或欧盟市售原研地舒单抗(PROLIA)。在第52周,接受PROLIA的受试者再次按1:1随机分配接受第三剂HLX14或PROLIA。本研究的共同主要终点为第52周腰椎BMD(LS-BMD)从基线变化的百分比(由中心影像评估)及基线至第26周的血清胶原蛋白C端肽(s-CTX)较基线改变百分比-时间曲线下面积 (AUEC0-26W)。次要终点包括腰椎BMD基线变化百分比(由研究者评估),股骨颈及全髋部BMD的百分比变化(由中心影像及研究者评估),骨折率,s-CTX及血清N-末端I型前胶原肽(s-P1NP)的相对变化百分比,安全性,药代动力学(PK),和免疫原性。
在基线到第52周期间,HLX14组和PROLIA组的腰椎骨密度(LS-BMD)均有所增加。通过中央影像评估,从基线到第52周的平均(标准偏差)百分比变化分别为HLX14组6.10%(3.95%),PROLIA组5.90%(3.83%)。两组之间的调整平均差异为0.23%(90%置信区间[CI]:−0.36%,0.83%;95% CI:−0.48%,0.95%)。90%置信区间和95%置信区间均落在预设的等效性界限−1.45%到1.45%之间, 证明了两组疗效等效性。对于主要药效动力学(PD)终点,HLX14组和PROLIA组的AUEC0-26W几何均值(CVb%)分别为14075.1253(17.3%)day*%inhibition,13883.3613(17.9%)day*%inhibition。HLX14组和PROLIA组AUEC0-26W的几何均值比为1.01(90% CI:0.99, 1.04;95% CI:0.98, 1.05)。几何均值比的90%和95%置信区间均落在预设的等效性界限0.8到1.25之间,证明两组PD等效性。因此,本研究的共同主要终点已达到。所有敏感性、补充性和亚组分析结果也与主要分析一致。所有次要疗效终点的结果进一步支持了HLX14和欧盟市售原研地舒单抗(PROLIA)等效。

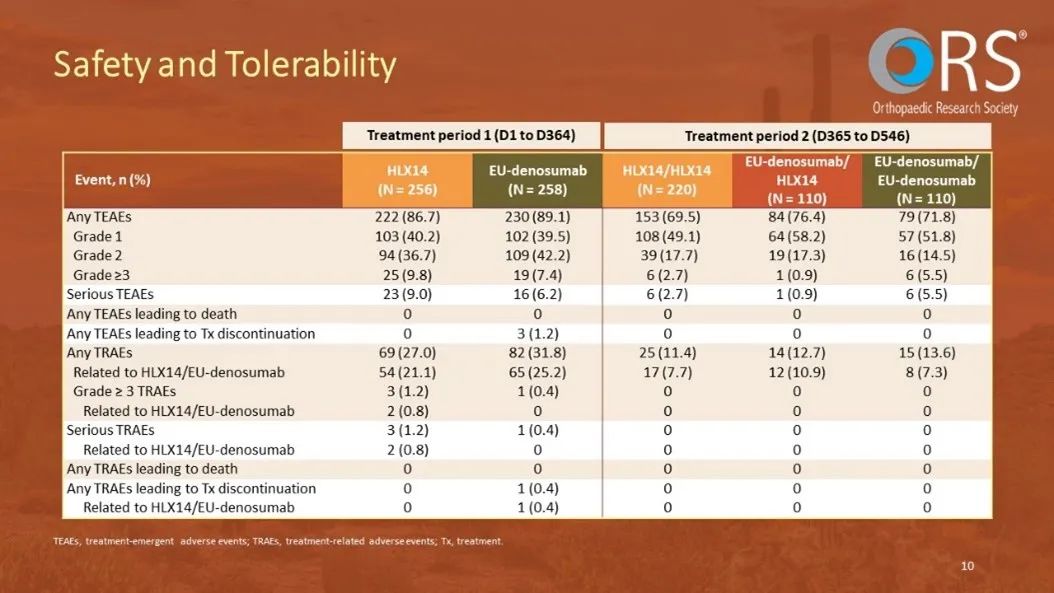
在基线到第52周期间,两组的安全性结果相当;HLX14组出现了69例(27.0%)与治疗相关的不良事件(TRAEs),而PROLIA组出现了82例(31.8%)。从第52周到第78周,PROLIA/HLX14、PROLIA/PROLIA、HLX14/HLX14各组之间的安全性特征仍相似;PROLIA/HLX14组报告了14例(12.7%)TRAEs,PROLIA/ PROLIA组报告了15例(13.6%),HLX14/HLX14组报告了25例(11.4%)。总体而言,安全性评估未发现HLX14和PROLIA之间存在显著差异,即使在单次治疗药物转换之后也是如此。各组在单次药物转换前后的抗药抗体和中和抗体的发生率也相当。